|
Organic Reactions |
How and Why Reactions "Go"
|
|
|
|
|
1 Different Kinds of Reactions |
We NEED some language to distinguish among various kinds of organic reactions
1. Addition Reactions (introduced in Alkenes)

� H and Br are added to the C=C double bond of the alkene
2. Substitution Reactions (introduced in Alkyl Halides)

� Br is substituted for OH in the alkyl halide
3. Elimination Reactions (introduced in Alkyl Halides)

� H and Br are eliminated from the alkyl bromide
� the HO� is required to "make" this particular elimination reaction "go"
4. Rearrangement Reactions (introduced later in Alkynes, Alcohols etc.)
![]()
� the reactant and product are structural isomers, the bonds are rearranged� the H3O+ is required to "help" this particular rearrangement reaction "go", it is a catalyst
5. Protonation/Deprotonation Reactions

� A proton is transferred from the acid (H-Cl) to the ketone
� the ketone becomes protonated
� the H-Cl protonates, it becomes deprotonated
|
2 Br�nsted Acidity |
� Acid/Base reactions play a DOMINANT ROLE in organic chemistry, we need to understand this VERY WELL
� A Br�nsted acid DONATES a proton
� A Br�nsted base ACCEPTS a proton
Example from general chemistry

� WHY does an acid react with a base to give a salt plus water?
A. Because in doing so it MAKES A NEW BOND, and lowers the energies of a pair of electrons
Example from organic example

� hydrogen moves from the acid to the base, but leaves its electron "behind" (is given to the oxygen). A hydrogen with no electron is a proton, therefore a proton is transferred from the acid to the base.
� This reaction makes a strong new bond and replaces a localized non-bonding pair with a resonance stabilized pair, even though an anion is formed, the electrons are stabilized by resonance, which is usually the most important factor for non-bonding electrons
|
2.1 Br�nsted Acid Strength |
Measured in terms of pKa (usually in water)

� Bronsted acidity involves HETEROLYTIC cleavage of a bond to hydrogen, with liberation of a proton
� stronger Bronsted acids have smaller pKa values
� this description ignores the role of solvation, but it is sufficient for now and helps us to understand Bronsted acidity and basicity within the context of bonding that we are using to describe most of the chemistry in this course
|
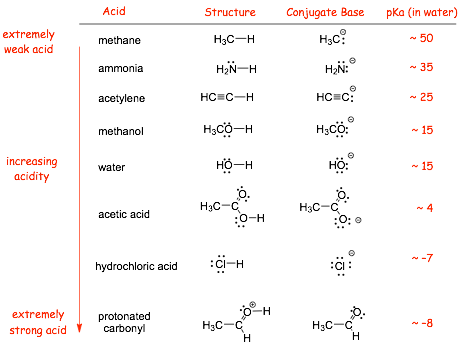
2.2 Range of Acidities in Organic Chemistry |
� We will encounter acids with a VERY WIDE RANGE of acidities
� We will consider the acidity of some protons that are so weakly acidic that they would not normally be considered as acids
� We will also encounter some organic species that are a lot stronger acids than sulfuric acid (although they will not normally be very long lived, especially in water!)
� REMEMBER, the pKa scale is logarithmic! So, an acid with a pKa of 10 is not ten times stronger than an acid with pKa of 20, it is 10 ORDERS OF MAGNITUDE STRONGER!!
Some Example Acids We will Meet
|
2.3 Factors Controlling Br�nsted Acidity |
� Absolute acidity (pKa) is difficult to predict, relative acidity is much easier!!
� Just as when we considered BDE as a simple chemical reaction, we need to consider BOTH the electrons that are involved in the REACTANT AND THE PRODICT, i.e. BEFORE and AFTER bond heterolysis
When Considering the Energetics of ANY CHEMICAL Reaction: We need to consider BOTH the energy of the electrons in the REACTANT (where we "start") and also the PRODUCT (where we "end")
� A stronger acid "wants" to go from left to right more than a weaker acid, either because the energy of the electrons in the bond in the REACTANT acid are high, or because the electrons in the PRODUCT conjugate base anion are low
![]()
� note, this is HETEROLYTIC BOND CLEAVAGE (this is NOT given the bond dissociation energy)
� A STRONGER ACID has a less reactive conjugate base anion, thus WEAKER BASE
� in general, an acid will be stronger if the electrons in the bond are high in energy, i.e. if the bond is weak
� MOST OF THE TIME, an acid will be stronger if the energy of the now non-bonding electrons on the conjugate base (usually on an ANION) are lower in energy, e.g. if B is more electronegative than A in the example above, the conjugate base B is easier to form, B-H is the stronger acid
� PROBLEM, these two effects SOMETIMES OPPOSE, how to know which one "wins"
|
2.4 Factors That Determine Bronsted Acidity ENERGIES OF ELECTRONS IN THE REACTANTS |
� In contrast to the BDE trends we looked at above, most of the trends on Bronsted acidity can be understood in terms of the energy of the electrons in the reactants, not the product, however, there is one very strong effect on Bronsted acidity that is determined by the energy of the electrons in the REACTANT
� Acidity increases with increasing atomic size going down a group in the periodic table.
Example
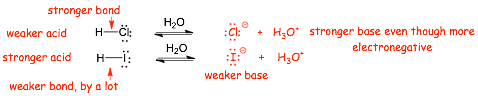
� H-I is a stronger acid than H-Cl
� Chlorine is smaller than iodine, the H-Cl bond is much stronger than the H-I bond because of poor orbital overlap between the very small hydrogen and very large iodine
� NOTE that we are using the same arguments we used for homolytic BDE here, these same effects influence the HETEROLYTIC cleavage associated with Bronsted acidity
� The iodide conjugate base anion is the weaker base because it makes weaker bonds when it reacts as a base
� The STRONGER ACID has the WEAKER CONJUGATE BASE
� The STRONGER ACID is the one with the WEAKER BOND
� NOTE: we could have tried to argue that the chloride anion should have been the weaker base, because chlorine is more electronegative than iodine and the energy of the electrons in the conjugate base anion should be lower, however, we need to consider BOTH the reactant and product, and it turns out that the bond strength effect "wins"
� There are often competing arguments when considering Bronsted acidity, the energy of the electrons in the ACID REACTANT wins here. Atomic size effects are often a dominant effect in organic chemistry.
|
2.5 Factors That Determine Bronsted Acidity ENERGIES OF ELECTRONS IN THE PRODUCTS |
� MOST of the trends on Bronsted acidity can be understood in terms of the energy of the electrons in conjugate bases anion PRODUCTS
1. Influence of Resonance on the stability of the conjugate base anion products
� Acidity increases with increasing resonance stabilization of the non-bonding electrons in the conjugate base anion. Resonance decreases the energy of the electrons in the conjugate anion base, making the anion easier to form, the acid is stronger. Resonance stability effects in the conjugate base anion usually dominate over all other possible factors.
Example:
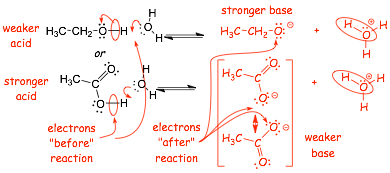
� the energy of the electrons are lower in the second conjugate base anion due to resonance stabilization
� the second reaction "wants" to go from left to right more than the first, the second structure is the stronger acid
� resonance stabilization in the anion and bond strength contribute to making the second structure more acidic
� the second conjugate base is the weaker base, it is the more stable anion
� The STRONGER ACID has the WEAKER CONJUGATE BASE
� The STRONGER ACID has the conjugate base with the LOWER ENERGY ELECTRONS
� there are no competing factors, this is straightforward, the lower energy electrons in the base argument works well
2. Influence of Electronegativity on the stability of the conjugate base anion products
� Acidity increases going from left to right across the periodic table. The stabilizing effect of increased electronegativity of the atom carrying the negative charge in the conjugate base anion is more important than increasing bond strength.
Example:
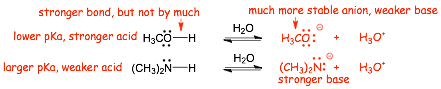
� The O-H bond is slightly stronger than the N-H bond, HOWEVER, the non-bonding electrons in the conjugate base anion are much more stabilized (lower in energy) on the more electronegative oxygen in the first structure, this effect on the PRODUCT is more important than the bond strength
� The STRONGER ACID has the WEAKER CONJUGATE BASE
� The STRONGER ACID has the conjugate base with the LOWER ENERGY ELECTRONS
� the lower electron energy in the base argument "wins" over the COMPETING stronger bond effect
3. Influence of Hybridization on the stability of the conjugate base anion products
� Acidity increases with increasing s character of the hybridization of the atomic orbital in the conjugate base atom that holds the non-bonding electrons, since increased stability of the electrons in the smaller A.O. in the conjugate base anion is more important than increasing bond strength. Hybridization effects are also often very important.
Example:
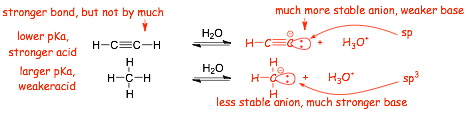
� the C-H bond in the first structure is stronger due to overlap of an sp AO on carbon compared to sp3., HOWEVER, the non-bonding electrons in the conjugate base anion are much more stabilized (the electrons are lower in energy) in the smaller and lower energy sp atomic orbital in the first structure, this effect is more important than the bond strength
� The STRONGER ACID has the WEAKER CONJUGATE BASE
� The STRONGER ACID has the conjugate base with the LOWER ENERGY ELECTRONS
� the lower electron energy in the base argument "wins" over the COMPETING stronger bond effect
SUMMARY: THE STRONGER ACID HAS THE LOWER ENERGY ELECTRONS IN THE CONJUGATE BASE ANION, EXCEPT WHEN COMPARING ACIDS GOING DOWN THE PERIODIC TABLE
We have explained acidity in terms of electron energies, energies are related to thermodynamics, and so we have made thermodynamic arguments. This makes sense and connects to our understanding of acidity from general chemistry. Acid strength is related to pKa, which is related to the equilibrium constant for dissociation, and the equilibrium constant is determined by thermodynamics, the energy differences between the different sides of the equilibrium
|
2.6 Substituent Effects and Bronsted Acidity |
� We will often be concerned with the effects of "substituents" on organic reactivity
� Substituents can affect the energies of electrons directly via the inductive, resonance and related effects. We call these electronic effects, the inductive effect is discussed in this section
� Substituents may also affect electron energies less directly via steric effects that we will discuss later
The Inductive Effect:
� The INDUCTIVE EFFECT is the lowering of electron energy due to electronegative atoms (as a consequence of the large positive charge on the nucleus)
� ALREADY SEEN, inductive effect is the origin of bond dipole moments, more electronegative elements �attracts� electrons by lowering their energy
� The inductive effect acts �through� sigma-bonds, and decreases rapidly as the number of bonds between the electronegative atom and the site of interest increases
� WIthin the context of Bronsted acidity, the inductive effect lowers the energies of the electrons in the anion conjugate bases, lowering energy makes the anions more stable, easier to form, increasing Bronsted acidity
Example: Which is the strongest and which is the weakest Bronsted acid?
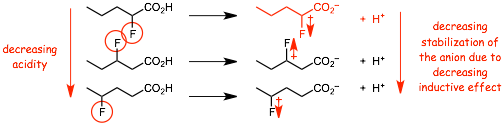
� The inductive effect of the fluorine substituent thus lowers the energies of the non-bonding electrons on the negatively charged oxygens in the conjugate base anion
� The inductive effect of the fluorine decreases rapidly with distance, it gets smaller very quickly with increasing numbers if intervening sigma-bonds
� The STRONGER ACID has the WEAKER CONJUGATE BASE
|
2.7 Predicting Br�nsted Acid/Base Reactions Using pKa |
� Easy when you know the pKa of each acid involved�
Example 1:
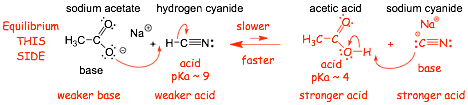
� the stronger acid protonates faster, equilibrium on the LEFT, the reaction �goes� from right to left
� "protonation" of the cyanide anion with acetic acid (the stronger acid) is exothermic and faster
� "protonation" of acetate with hydrogen cyanide (the weaker acid) is endothermic and slower
Example 2:
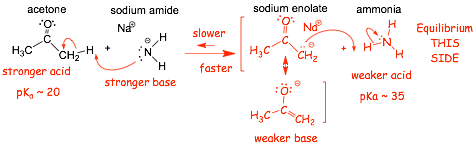
� proton transfer from the stronger acid is faster, thus equilibrium lies to the right
� "deprotonation" of acetone (the stronger acid) using sodium amide is exothermic and faster
� "deprotonation of ammonia (the weaker acid) using sodium enolate is endothermic and slower
� the reaction �goes� from left to right
2.8 Predicting Br�nsted Acid/Base Reactions Using Electron Energy Arguments |
� We can use our knowledge of relative acidities to predict the outcome of Bronsted acid/base reactions even if we don't know the relevant pKas.
Example, On which side will this equilibrium lie?? (which is stronger acid??)
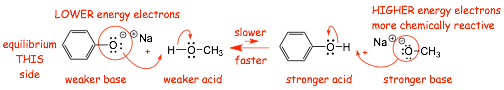
� MORE stable anion on the left (resonance stabilized), lower energy electrons, less reactive, WEAKER BASE
� Conjugate acid of the weaker base is the stronger acid (formation of the anion is easier)
� LESS stable anion on the right (NOT resonance stabilized), higher energy electrons, STRONGER BASE
� Conjugate acid of the stronger base is thus the weaker acid (formation of the anion is harder)
� The reaction of the stronger acid with the stronger base is FASTER, the reaction of the weaker acid with the weaker base is SLOWER, equilibrium lies on the left
Example: will water protonate methyl lithium?
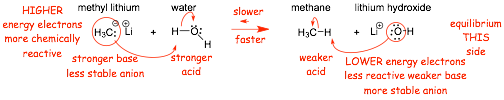
� the stronger acid reacts with the stronger base faster
� the weaker acid reacts with the weaker base slower
� Equilibrium lies to the RIGHT, the reaction �goes� to the right
� so, the answer to the question above is YES, water will protonate CH3Li (explosively!!!!!!!!!)
|
3 Lewis Acid/Base Reactions |
� THE MOST IMPORTANT CONCEPT IN THE ENTIRE COURSE!
Question?: Why DOES acid + base give salt + water?

Answer: Because this gets TWO electrons into a bond, thus lowering their energy
Actually, in water it is quite a bit more complicated than this because of ionic solvation effects, but the point of the question is clear, we can UNDERSTAND acid/base reactions in terms of the fundamental electron energy effects that we have been talking about during this course
G. N. Lewis proposed that the most significant thing about the above reaction is NOT that a proton was transferred from the acid to the base, but:
� the BASE DONATED two electrons to make the new bond
� the ACID ACCEPTED two electrons to make the new bond
This is Lewis's (broader) definition of an acid and a base
Example 1:
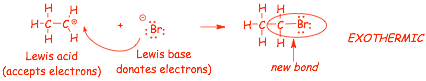
� this reaction "goes" from left to right because it gets 2 electrons into a bond, lowering their energy, the reaction is exothermic
Example 2:
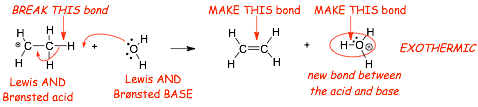
� this reaction "goes" because although 1 bond is broken, 2 are formed
� the acid transfers a proton to the base, this is thus BOTH a Lewis and a Br�nsted acid/base reaction
IMPORTANT � ALL Br�nsted acids/bases are also Lewis acids/bases
� NOT all Lewis acids/bases are Br�nsted acids/bases
Example 3: not all acid/base reactions have to be exothermic:
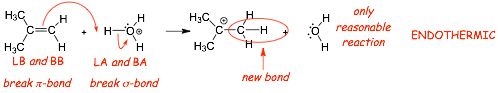
� THIS reaction breaks TWO bonds in the reactants (the pi-bond in the alkene and the H-O sigma-bond in the H3O+ and only makes one new C-H sigma-bond, bonds bonds are broken than are made
� the products of this reaction are thus higher in energy than the reactants (the reaction is endothermic)
� nevertheless, we will see later that reactions such as these are important
� Lewis acid/base reactions can be "complete" single step reactions in their own right as in the examples above, or they can be "parts" of more complex reactions which have multiple steps (see later).
|
3.1 Predicting Lewis Acid/Base Reactions |
Here is where we learn to how to avoid memorizing every reaction in organic chemistry!!!
give the product of the following reaction:
� formaldehyde could give electrons (Lewis base) or accept electrons (Lewis acid)\
� hydride anion can only reasonably give electrons, thus it must be the Lewis base
� which atom on formaldehyde should it make a new bond to? Let's try and see....
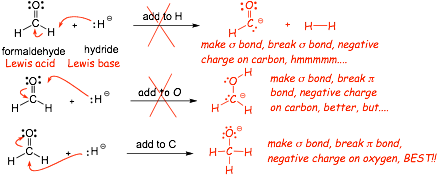
give the product of the following reaction:

� the non-bonding electrons on nitrogen are higher in energy (more reactive) than those on oxygen because it is less electronegative
� this is the most reasonable reaction for this pair
4 Curved Arrow-Pushing |
� We need to return to curved arrow-pushing since it is a critical part of the organic chemistry LANGUAGE
� curved arrows represent the movement of electrons (only)
� the arrow "starts" where the electrons come from, "ends" where they go to
Examples:
![]()
� draw a dotted line to show the new bond if it helps you

� the arrow shows that how the electrons are used to make the bond, NOT how the atoms "move"
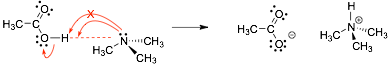
� The arrow starts at the electrons, and ends "in" the new bond, NOT at the H, since the electrons are not given to the H.
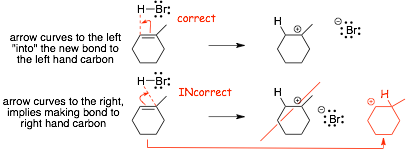
� the arrow shows bond formation AND implies exactly which atoms are involved in formation of the new bond, the "curved right" arrow above implies making a bond from H to the carbon on the right in the C=C bond, but the product shows the new bond being made to the carbon on the left, a "right curved" arrow would be incorrect in this case.
Visualize Curved Arrow Pushing
|
5 How and Why Chemical Reactions �Go�? |
|
5.1 The ANATOMY of an organic reaction equation |
� Organic chemists are often LAZY, organic reactions written in equation form are often not completely balanced, they also focus almost exclusively on the organic structures and not the other chemicals that are required to make the reaction go

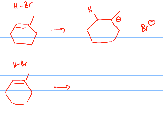
� The reagents/conditions summarize what chemicals and reaction conditions are required to make the reaction go, in case it is conversion of an alkene into an alkyl bromide
� There are TWO alkyl bromide products of this reaction, the MAJOR product is usually the DESIRED product of the reaction, any other reaction products are often not wanted and are called SIDE PRODUCTS
� We will return to why there are more than one product of this reaction and why one is the major later
� Organic reactions are usually performed either in the LIQUID STATE or in a SOLVENT, in order to allow the molecules to undergo the collisions required for reaction, the solvent may not be specified if it is not important what it is, in this case the solvent is carbon tetrachloride (CCl4) which is inert, meaning it does nto get involved in the reaction, it is only there to solvent the alkene and the H-Br
|
5.2 Why do Chemical Reactions "Go"? |
Q. How do we know when we mix to chemicals together that they will actually react?
A. If the reaction is favorable energetically AND kinetically, here we look at the problem from the thermodynamic perspective (the kinetic perspective is coming later)
� Consider the following reaction, will it go from left to right?

� In principle, ALL chemical reactions are reversible (not just acid/base)
� thus, this reaction is predicted to "GO" from left to right, not the other way round, because the GIBBS FREE ENERGY of products on the right are lower than the GIBBS FREE ENERGY of the reactants on the left - the reaction from left to right is EXERGONIC (there is a negative free energy change from left to right)
![]()
What determines reaction (Gibbs) free energy? Enthalpy and Entropy
![]()
� for organic reactions it is the enthalpy term usually dominates
� the enthalpy term is given mainly by the energy of electrons (sometimes solvation)
� when the enthalpy term is small, the entropy can determine the reaction, but this is less common
� So, most of the time the enthalpy/energy of the electrons allows us to understand the reaction, which is good because electron energy is something that we know about!
Summary:
� Reactions "go" when they are exergonic (negative reaction free energy)
� enthalpy changes are usually more important than entropy changes (we will point out the cases where entropy does play a role as we proceed)
� enthalpy is related to electron energies
� electron energies in bonds are measured in terms of Bond dissociation energies
Important Terms to Understand
an exergonic reaction has a negative free energy
an endergonic reaction has a positive free energy
an exothermic reaction has a negative enthalpy, energy comes OUT of the reaction, it gets hot!
an endothermic reaction has a positive enthalphy, energy goes IN to the reaction, it feels cold!
|
5.3 Calculating Enthalpy Change Using Bond Dissociation Energies |
� Reaction ENTHALPY is defined as:
![]()
� BUT, we don't really KNOW the enthalpies of either the reactant OR the product, but enthalpies are related to electron energies, which in the context of reactions are related to bond dissociate energies
� And so we can work out reaction enthalpies A DIFFERENT WAY, not using the enthalpies themselves but INDIRECTLY, using bond dissociation energies
HOW IT WORKS:
LOOK at the energies of the bonds that are made and broken for the H-Br addition to alkene reaction
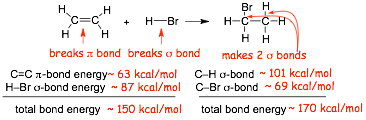
� Overall weaker bonds are converted into overall stronger bonds
� Specifically, one WEAKER pi-bond and one STRONGER sigma-bond are converted into TWO STRONGER sigma-bonds
� Energy has to go "into" the reactant molecules to break bonds, this is an energy (enthalpy) cost
� Energy comes out of the product molecules when bonds are made, this is an energy (enthalpy) release
� The reaction above "releases" more energy than it "costs" to do the reaction
� The electron energy DECREASES because stronger bonds are formed, therefore the reaction is EXOTHERMIC (electron energy ~ enthalpy)
![]()
� The absolute VALUE of the reaction exothermicity can be calculated from the difference in the bond dissociation energies
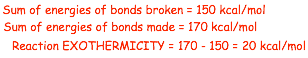
� The reaction ENTHALPY is equal to the reaction exothermicity (or endothermicity), because this reaction is exothermic, the reaction enthalpy must be negative (by definition):
![]()
SUMMARY: REACTION ENTHALPIES AND BDE (how to do it):
1) Decide if the reaction is exothermic or endothermic, do thus by summing the bond dissociation energies if necessary, sum of bonds broken versus sum of bonds made
2) If overall stronger bonds are made then the electron energy decreases and the reaction is exothermic, if weaker bonds are made then the reaction is endothermic
3) The absolute exothermicity or endothermicity is given by the difference in the bond dissociation energies
4) The reaction enthalpy is negative if exothermic or positive is endothermic, and the reaction enthalpy is quantitatively the same as the exothermicity or endothermicity
OR�.
� Determine the difference between the sums of the energies of the bonds that are made/broken and use COMMON SENSE to decide if this difference corresponds to a POSITIVE or NEGATIVE reaction enthalpy
|
5.4 Reaction Mechanisms |
� describes HOW a reaction occurs, the order in which bonds are made and broken
� describes which intermediates are involved
� is the key to minimizing memorization in organic chemistry!
Consider the following example:
the reaction
![]()
� This reaction is EXERGONIC and EXOTHERMIC
� It is exothermic because 1 sigma bond and 1 pi-bond are broken, but 2 stronger sigma-bonds are made
� Quantitatively because of the enthalpy calculation in the previous section of the notes
the mechanism
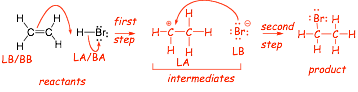
� this mechanism consists of two consecutive Lewis acid/base reactions
� the first step forms intermediates from the reactants, the second step forms the final product
� we will use the abbreviations LA/LB to represent Lewis acids/base and BB/LB to represent Bronsted acids/bases
|
5.5 Some Important Intermediates |
Before we go deeper into mechanisms we first need to take a short detour to look at some common intermediates in organic reactions
Carbocations (more correctly, carbenium ions)


� hyperconjugation "donates" electrons from the methyl group to the positively charge carbon, which delocalizes electrons, they "see" more nuclei, lowers electron energy, also "spreads out" the positive charge, thus the ethyl cation is more stable (less reactive) than the methyl cation.
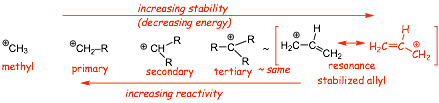
�
increasing stability from left to right as the cation is increasingly
stabilized by hyperconjugation, and ultimately, true resonance.
Radicals (already seen!)

� hyperconjugation can still delocalize one electron, smaller decrease in electron energy
� increasing stability from left to right as the radical is increasingly stabilized by hyperconjugation, and ultimately, true resonance
Carbanions
![]()

� this time electron REPULSION INCREASES the energy of the non-bonding electrons on the negatively charged carbon, any "attempt" at hyperconjugation actually results in an ANTI-BONDING interaction

� decreasing stability from left to right as the radical is destabilized by electron repulsion, however true resonance in the ally anion still "wins", since it delocalizes the electrons without an ANTI-BONDING interaction
|
5.6 Kinetics and Reaction Energy Diagrams |
� The thermodynamic description of a reaction discussed above indicates whether a reaction is possible, but says nothing about how FAST it might occur, a reaction that is favorable thermodynamically ( is predicted to "go), might be too slow to actually "go" in a reasonable timescale
� There is a CONNECTION between reaction thermodynamics and kinetics
Example: The reaction below proceeds via two (mechanistic) steps
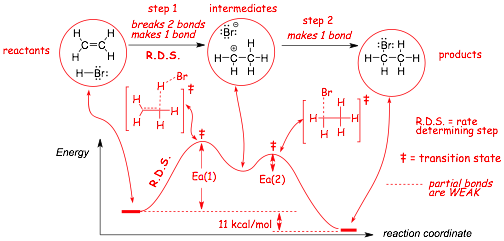
� This REACTION ENERGY DIAGRAM shows how the energy of the entire system changes during the course of the reaction
� The REACTION COORDINATE is an aggregate measure of all of the bond lengths and angles that have to change in order to go smoothly form the reactants to the intermediates and from the intermediates to the products
� the energy of the INTERMEDIATES are higher than either the reactants or the products, because they have one less bond, two bonds are broken and only 1 bond is made to form the intermediates
� the reaction proceeds via the LOWEST FREE ENERGY path, given by the reaction coordinate
� the reaction is overall exothermic because 1 sigma and 1 pi bond are converted into 2 stronger sigma bonds
Visualize Understanding Reaction Energy Diagrams
� Between the reactants and the intermediates is a TRANSITION STATE (�), which has PARTIAL BONDS that are not fully formed, which is why it is high in energy
� There is a high energy TRANSITION STATE associated with each step of the mechanism in any organic reaction
� Transition states are at ENERGY MAXIUMA, the definition of an intermediate is that it is in an energy MINIMUM, even if it is only a shallow energy minimum
� Each react step MUST GO OVER THE TRANSITION STATE, the energy required to reach the transition state is the ACTIVATION ENERGY (Ea).
� the RATE CONSTANT (k) for each reaction step is related to the size of its ACTIVATION ENERGY (Ea)
k = A exp(-Ea/RT)
� thus, a smaller Ea results in a larger k, i.e. a faster rate
� a larger Ea results in a smaller k, i.e. a slower rate
� MOST ORGANIC REACTIONS PROCEED VIA THE SMALLEST ACTIVATION ENERGY BECAUSE THIS IS FASTEST, MOST ORGANIC REACTIONS ARE KINETICALLY CONTROLLED!
� In this example above, Ea(1) > Ea(2), step 1 is thus slower and is the RATE DETERMINING STEP (R.D.S.)
Example Problem: which of the following two reactions A or B is faster??
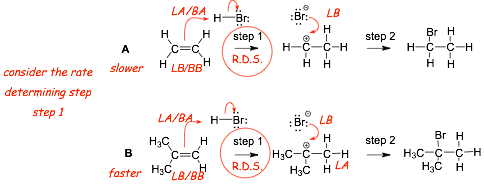
� When considering overall reaction arte we need to focus on the RATE DETERMINING STEP
� The rate determining step in A involves formation of a higher energy, less stable primary cation
� The rate determining step in B involves formation of a lower energy, more stable tertiarty cation
� The rate determining step in B is faster than that in A, thus reaction B is overall faster than reaction A
The Hammond Postulate:
All other things being equal (i.e. for similar or related reactions ONLY)
� With INCREASING EXOTHERMICITY, reactions will have earlier, lower energy transition states (�), and will be increasingly FASTER
� With INCREASING ENDOTHERMICITY, reactions have later, higher energy transition states (�), and will be increasingly SLOWER
The Reactivity/Selectivity Principle:
� For fast reacting intermediates such as radicals and cations, the reactivity/selectivity principle often applies
� The difference in reaction rates gets smaller as the reactions get faster (as they approach the fastest possible reactions they can't get any faster and can't get any different), i.e the reactions become LESS SELECTIVE
� The difference in reaction rates gets larger as the reactions get slower, i.e. the reactions become MORE SELECTIVE
The reactivity/selectivity principle turns out to be useful only really for very fast reactions of very reactive intermediates such as radicals, cations etc., it sometimes fails for other slower reactions.
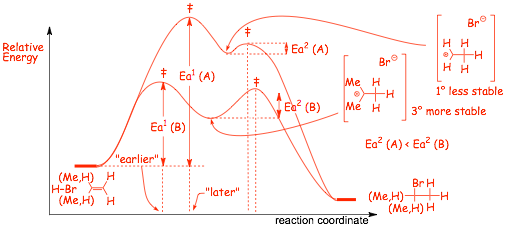
� The rate determining step, step 1, for reaction (A) is more endothermic than the same step for reaction (B)
� The more endothermic step has the LARGER Ea and the LATER transition state, Ea1(A) > Ea1(B)
� The more endothermic step also has a LATER transition state
� The first step in reaction A is SLOWER, because the intermediate cation is less stable
� The second step for reaction (A) is more exothermic than the second step for reaction (B)
� The more exothermic step has a SMALLER Ea, Ea2(A) < Ea2(B), and an earlier transition state
� The second step in (A) is faster because the intermediate is less stable, it has an EARLIER transition state
� Reactions that FORM less stable intermediates are SLOWER, Reactions OF less stable intermediates are faster
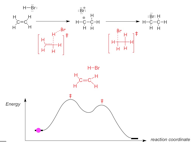
Reaction Energy Diagram for a Bronsted Acid/Base Reaction
The reaction:

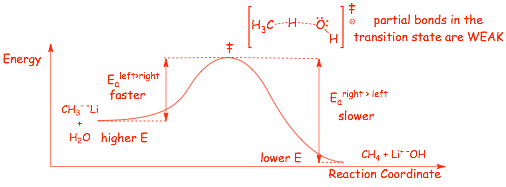
� The forward and reverse reactions MUST proceed via the same transition state: this is the principle of microscopic reversibility
� in the TRANSITION STATE, the distances between the atoms involved in bond making/breaking is large, the partial (dashed) binds are this WEAK, which is why the transition state is highest in energy
� the transition state is CHARGED (negative charge), but because it is not a Lewis structure we can't assign FORMAL charges to any specific atom, we indicate the charge "outside" the brackets