|
Alkyl Halides |
Substitution and Elimination
|
|
|
|
� halides mainly undergo SUBSTITUTION and ELIMINATION reactions

|
1 Nomenclature |
� Look for the longest chain that CONTAINS the MAXIMUM NUMBER of functional groups, in this case the halogen is the functional group and so even though the cyclohexane has more carbon atoms, the main chain is the two carbon ethane chain, the structure is named as a substituted alkyl bromide

|
2 Second Order Nucleophilic Substitution (SN2) Reaction |
SUBSTITUTION by making a new bond AT THE SAME TIME as breaking the old bond
� substitution requires a bond to be broken AND a new bond to be formed
� the LOWEST energy way of doing this (unless precluded by steric or other effects, see later) is to MAKE THE NEW BOND (getting some energy "back") at the same time as BREAKING THE OLD BOND, this is SN2
Example
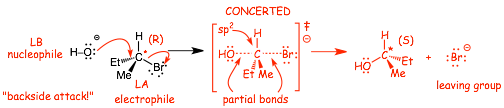
� This is fundamentally just a Lewis acid/base reaction of a kind we have seen previously, the Lewis base has the high energy chemically reactive electrons, which are used to make a new bond to the Lewis acid, and a stronger bond is formed (C-O in the example above) and a weaker bond is broken (C-Br above)
� HO� is the Lewis Base and Nucleophile
� the halide is the Lewis acid/electrophile
� the Br� anion is the Leaving Group
� This reaction "goes" because�.
1) a weaker bond is converted into a stronger bond
C�Br: B.D.E. ~ 65 kcal/mol (weaker bond)
HO�C: B.D.E. ~ 90 kcal/mol (stronger bond)
2) a stronger base (�OH) is converted into a weaker base (Br�)
3) in this way HIGHER energy electrons are converted into LOWER energy electrons
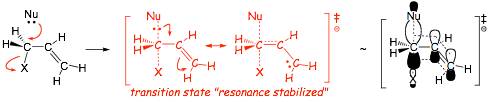
� Reactions in which all bonds are made and broken at the same time are called CONCERTED
� The reaction also proceeds with inversion (i.e. BACKSIDE ATTACK, think about an umbrella turning inside out in the wind!), called a Walden inversion. Often this will lead to a change in absolute configuration, i.e. R to S or vice versa, but not necessarily!

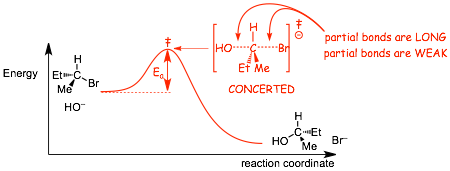
� even though the transition state apparently has one more bond than either the reactants or the products, the partial bonds are very LONG and thus WEAK, partial bonds thus have very high energy electrons which is why the transition state is higher in energy than either reactants or products (2 weak partial bonds add up to less than one real bond)
Why the Name Second Order Nucleophilic Substitution (SN2)??
S - Substitution reaction
N - Nucleophile does the substitution (like a Lewis base, but see below)
2- kinetically 2nd order, TWO molecules are involved in the rate determining step (the only step)
� The halide AND the nucleophile (2 molecules) are involved in the rate determining step and so the reaction rate depends upon the concentration of them both, the reactions is kinetically SECOND (2nd) order
![]()
� an increase in the concentration of EITHER or BOTH the nucleophile and halide results in a proportional increase in reaction rate, the rate depends upon the concentration of BOTH REACTANTS
What is a Nucleophile and How is it Different from/Same as a Base?
Definition of a BASE: Based on THERMODYNAMICS

� Lewis/Br�nsted base strength measured by size of Keq (thermodynamic definition)
� stronger base means stronger new bond means more exothermic reaction larger Keq
� weaker base means weaker new bond means less exothermic (or more endothermic) smaller Keq
Definition of a NUCLEOPHILE: Based on KINETICS
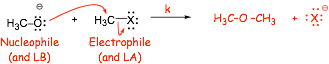
� Nucleophile strength measured by size of rate constant k (kinetic definition)
� stronger nucleophile means smaller Ea (stronger partial bonds), larger k, faster reaction rate
� weaker nucleophile means larger Ea (weaker partial bonds), smaller k, slower reaction rate
All nucelophiles are Lewis bases, the Hammond postulate says that strong bases should also be strong nucleophiles, and this is generally true, although we will meet a few important exceptions later�..
Lewis Base / Nucleophile (nucleus loving), donates electrons
Lewis Acid / Electrophile (electron loving), accepts electrons
Why are we concerned with kinetics now when we used to be only concerned with the acid/base understanding of reactivity? We have already seen that when there are competing reactions, the fastest one "wins" (e.g. the most stable intermediate is formed fastest), in other words MOST organic reactions are controlled by kinetics, their reactions are KINETICALLY CONTROLLED. For this reason it makes sense to start talking about nucleophiles and electrophiles, because their definition is based on kinetics. Of course, most strong nucleophiles react fast BECAUSE they are also strong bases and have very exothermic reactions (although there are some exceptions). We will use the terms nucleophile/electrophile and Lewis base/acid interchangeably.
Examples of SN2 Reactions; Give the major organic product in the following reactions
� we understand these SN2 reactions a simple Lewis acid/base processes
� identify the Lewis base/NUCLEOPHILE as the reactant with the high energy electrons
� the Lewis acid/nucleophile must react with the Lewis acid/ELECTROPHILE
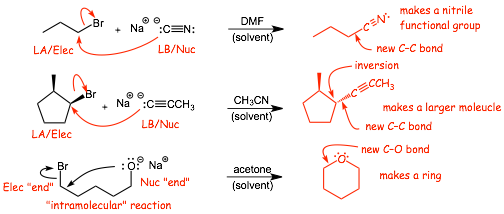
� IMPORTANT POINT: SN2 reactions are one of the most important ways of MAKING NEW BONDS, i.e. of transforming one organic molecule into another one
� Here we made a NEW FUNCTIONAL GROUP (nitrile), we made a new C-C bond (larger molecule), we also made a ring structure, we will use SN2 a LOT!
|
2.1 Factors Controlling SN2 Reactivity: Leaving Group Ability |
Good leaving groups:
� Are stable/less reactive as an anion
� Are generally weak bases (i.e. they have WEAK bonds to, for example, H atoms)
� Polarize the C�LG bond (and are polarizable to make strong partial bonds in transition state)
FOR EXAMPLE

� Leaving Group ability INCREASES going down the periodic, as ANION STABILITY increases
� The IODIDE ANION is stable and unreactive because it is a weak base, it makes WEAK BONDS to H
� The CHLORIDE ANION is LESS stable and is MORE reactive because it is somewhat stronger base, it makes somewhat STRONGER BONDS to H
Reaction Energy Diagrams: Two Equally correct ways of illustrating this effect
� Which of the following energy diagrams best illustrates why iodide is a better leaving group than bromide etc.?
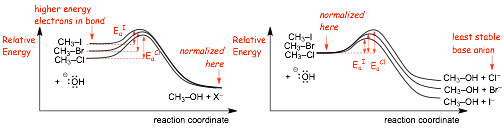
� BOTH of these diagrams are equally correct
� The diagram on the LEFT, the diagram shows the difference in the energies of the electrons on the bonds, the weaker (higher electron energy) bond is the most reactive (smallest Ea, largest reaction rate)
� The diagram on the RIGHT shows the difference in the stabilities of the anion leaving groups, the more stable the anion the smaller the Ea, the faster the reaction
� REMEMBER, the iodide anion is the most stable not because of particularly low electron energy in this case, but because if it reacts it can only make weak bonds because it is so large
|
2.2 Factors Controlling SN2 Reactivity: Solvent Effects |
� Nucleophile strength depends upon SOLVENT, this is something new for us
� Solvent effects on reactions are often DRAMATIC
� BUT, A complication: solvent effects are different for nucleophiles of different sizes (see below)
Polar PROTIC (hydrogen-bonding) Solvents: Mostly alcohols and water
� Polar protic solvents have STRONG intermolecular ion-dipole forces with dissolved anions in particular
� These ion-dipole interactions solvent anions strongly (think about ionic compounds dissolved in water)
� The STRONGER the IMF, the more solvated the ion in the solvent since the interactions with the solvent lower the total electron energy more than for comparable weaker intermolecular forces
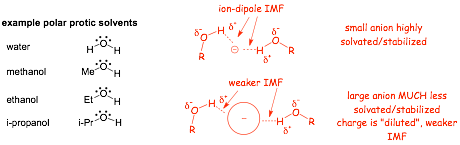
� Stronger solvation lowers electron energy and chemical reactivity, anions in particular tend to be MORE STABLE in polar protic solvents
� polar protic solvents solvate small anions VERY WELL, but solvate larger anions LESS WELL, transition states are LARGE, and are thus much less solvated
� there is often a LARGE solvation energy difference between reactants and the transition state, particularly when using small anionic nucleophiles in a polar protic solvent
� Intermolecular FORCE is actually an unfortunate term because force and energy are not the same thing, but it is not hard to understand that if an intermolecular force is strong it is more likely to lower the energy of the relevant electrons more
Polar APROTIC (NON-hydrogen-bonding) Solvents: There are Several, you need to know these
� Polar protic solvents have WEAKER intermolecular ion-dipole forces with dissolved ions
� The ion-dipole interactions certainly solvent and stabilize ions, but the H-bonding effect particularly on anions is MISSING
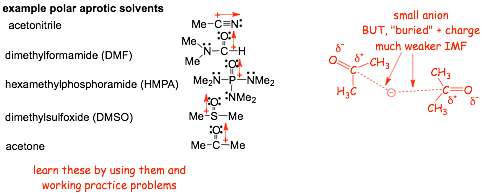
� There is a usually a SMALLER energy difference between reactants and the transition state when SN2 reactions are performed in polar APROTIC solvents, SN2 reactions in polar APROTIC solvents are usually faster than in polar protic solvents (but see further below)
� you NEED TO KNOW the polar APROTIC solvents, this is not easy because they are different structures, learn them by working with them
Medium Polarity or Non-Polar Solvents
�
These solvents should be GREAT for SN1 and SN2 reactions since they will
solvate the anions so poorly, but that is the problem, they solvate ions so
poorly that the reactants often simply don't dissolve in them!
� We do see these solvents quite often in organic reactions, in fact we have
seen carbon tetrachloride, CCl4) already, as an inert (non-reactive) solvent in
more than one reaction
� Some common low polarity solvents are summarized here but we will not use them much for Sn1/Sn2
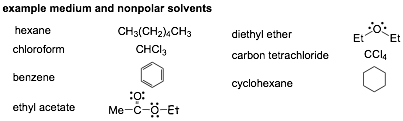
� Medium and in particular non-polar solvents are commonly used in organic chemistry, but not so much for SN2 reactions, since these often involve ionic reactants that will simply not dissolve in non-polar solvents
Example Solvent Effects

� Explain the difference in reaction rates using a reaction energy diagram
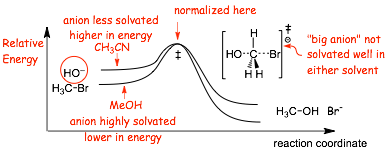
� here we have TWO reactions on one diagram. The ABSOLUTE energies of the two systems are very different, thus we need to NORMALIZE the energies and plot RELATIVE ENERGY.
� Where to normalize? In general we will want to emphasize the places where the energies are different and where they are similar. In this case the energies at the start and end are different due to large differences in anionic solvation, and the energies at the transition states are more similar due to small differences in solvation, thus we normalize at the transition state
|
2.3 Factors Controlling SN2 Reactivity: Nucleophilicity |
Good nucleophiles:
�Make strong partial bonds in the transition state, to decrease the transition state energy
Nucleophiles DONATE A PAIR OF ELECTRONS to make a new bond: identical to a Lewis base, BUT, nucleophilicity is based on reaction RATE, whereas basicity is based on reaction exothermicity
� Clearly, most good nucleophiles are also good Lewis bases, but there are some differences
Comparing same atom (charged versus non-charged)
� Anions make stronger nucleophiles than neutrals
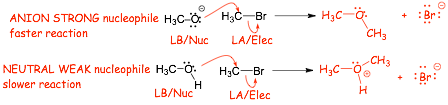
� NUCLOPHILICITY is the SAME as BASICITY here!
� higher energy electrons on negatively charged oxygen both react faster and have more exothermic reactions, are good Lewis bases AND good nucleophiles, this is true IN ALL SOLVENTS
Comparing similarly sized atoms (across the periodic table)
� Electronegative atoms make poor nucleophiles
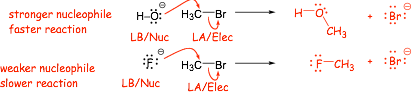
� NUCLOPHILICITY is the SAME as BASICITY here!
� lower energy electrons on electronegative fluorine react slower and have less exothermic reactions, are weaker Lewis bases AND less good nucleophiles, this is true IN ALL SOLVENTS
Comparing differently sized atoms (down the periodic table)
� A NEW CONCEPT. Larger atoms have more polarizable electrons, they do not have to "get so close" too make a bond, can make "longer" bonds, and thus can make stronger PARTIAL BONDS in the transition state
Example
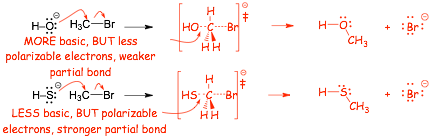
Question: So, which "wins", basicity or nucleophilicity because of the partial-bond strength?
Answer: Nucleophilicity depends upon SOLVENT, ugh!
Example: Compare these two reactions in a polar APROTIC solvent (or even gas phase!)
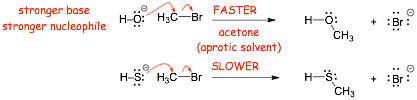
� The larger S makes stronger partial bonds, but the the stronger bonds in the product with O simply wins!
� NUCLOPHILICITY is the SAME as BASICITY here!

Example: Compare these two reactions in a polar PROTIC solvent

� NUCLOEPHILICITY and BASICITY are OPPOSITE HERE!

� the larger more polarizable sulfur anion forms stronger partial bonds in �, opposite to basicity
� AND, the larger sulfur anion is not solvated well (not stabilized) by the polar protic solvent, more reactive
� here nucleophilicity is OPPOSITE to basicity, solvation and polarization effects "win" over exothermicity
� to curves emphasize the difference in solvation of the nucleophilic ions
Nucleophilicity is ALMOST ALWAYS THE SAME as BASICITY, EXCEPT FOR�.
� larger anions are more nucleophilic than smaller anions, IN PROTIC solvents
going down the periodic table often makes things complicated!
|
2.4 Factors Controlling SN2 Reactivity: Molecular Orbitals and Steric Effects |
� Here we will use a more detailed form of Lewis acid/base theory that considers the important MOLECULAR ORBITALS, i.e. Frontier Molecular Orbital theory (FMO Theory)
� We need to make a BOND between two molecules
� Making bonds between ATOMS requires overlap of ATOMIC orbitals IN PHASE to generate a new BONDING molecular orbital
� Making a bond between MOLECULES requires overlapping MOLECULAR orbitals IN PHASE to generate a new BONDING molecular orbital
� F.M.O. theory looks at the overlap between MOLECULAR orbital with the highest energy electrons (the HOMO) of nucleophile, with the lowest energy MOLECULAR orbital of the electrophile (the LUMO)
� The HOMO is where the reactive Lewis basic electrons "are", the anti-bonding LUMO is the only orbital that the electrons can "go to" in the electrophile, all of the bonding orbitals are full of electrons
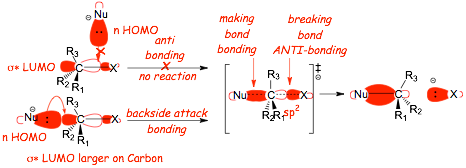
� This provides the BEST explanation for "backside attack", HOMO/LUMO overlap best at the carbon "end" of the halide LUMO
� for this reason, reaction suffers "steric hindrance" when R1, R2, R3 are large
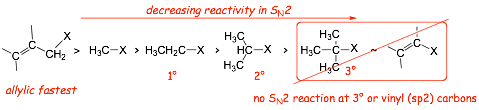
� SN2 reactions get slower with increasing steric hindrance at the backside of the carbon of the electrophile
� To the extent that there is NO SN2 REACTION at a TERTIARY halide
� SN2 reactions at METHYL and ALLYLIC carbons are particularly facile (see below)
There is NO SN2 at a tertiary or vinyl carbons because the nucleophile cannot get close enough to form reasonable partial bonds in the transition state due to a steric effect at the other alkyl substituents on the C atoms

� The vinyl C(sp2)-X sigma* orbital is smaller than a C(sp3)-X sigma* orbital, weakens any potential partial bond
� The vinyl C(sp2)-X bond is stronger than a corresponding C(sp3)-X bond
SN2 at the allylic position is FASTEST because the transition state is resonance stabilized, lowering the energy of the electrons in the transition state
� A lower energy transition state means a smaller activation energy which results in a faster reaction��
� ANOTHER example of a steric effect is provided by the nucleophile

� a good example of difference between basicity and nucleophilicity. t-butoxide is a very strong base (it forms a strong bond to a small proton), but a weak nucleophile, since it can't form a strong partial bond in the transition state with carbon. SN2 is not possible using the t-butoxide anion
|
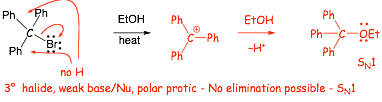
3 First Order Nucleophilic Substitution (SN1) Reaction |
� What happens if we try to do an SN2 reaction with a very weak (e.g. neutral) nucleophile Lewis base?

� here we have a nucleophilic substitution reaction BUT......
� we have a 3� halide which is a weak electrophile (backside attack is not possible), can't do SN2
� H3COH is a weak nucleophile (no negative charge on the oxygen), shouldn't do SN2
� H3COH is also a PROTIC solvent, which should be slow for SN2
� here the solvent "helps" to break the C�Br bond, the reaction is a solvolysis reaction (lysis - bond breaking)
We need a new substitution MECHANISM to account for this: The SN1 Mechanism
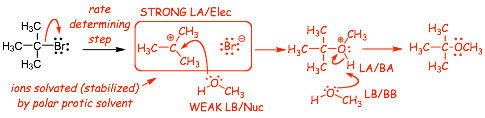
� although the alcohol is a weak LB/Nucleophile, the first cation intermediate is a STRONG LA/Electrophile, and so nucleophilic addition at this step in the mechanism is fast
� the SN1 reaction requires a polar protic solvent to stabilize the ionic (cation and halide) intermediates
� usually requires heat (energy) to break the C�X bond unimolecularly
� ONLY the halide (not the nucleophile) involved in the R.D.S., thus SN1 (1 means only 1 reactant in the R.D.S.)
� requires a stable intermediate cation, NO SN1 for methyl or primary halides
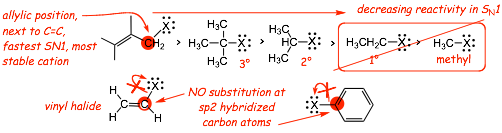
� No SN1 (OR SN2) at sp2 hybridized carbons, the C-X bond is too strong and the cations are too unstable
� In general, SN1 will always occur in preference to SN1 since this makes a bond at the same time the bond is broken, unless SN2 is impossible (e.g. at a 3� carbon)
|
3.1 Stereochemistry of SN1 Reactions: Racemization (?) |
Example
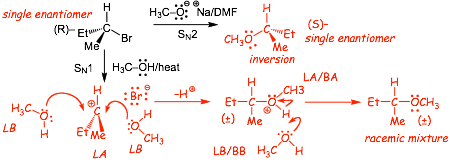
� We expect racemization, or at least some loss of stereochemistry for SN1 compared to SN2
� depending upon conditions/reactants, attack on the same side as the leaving group may be hindered, resulting in a slight excess of the inversion product
� in reality, however, it is not easy to predict exactly how much stereochemistry will be lost, and so we will use the "rule" in this course that if the reaction goes via SN1 we will assume that racemization always occurs
|
3.2 Cation Rearrangements in SN1 Reactions |
Example
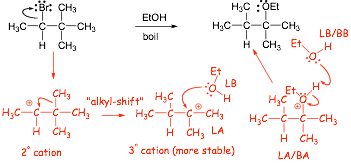
� Once the cation is made it will react the same as any other cation, and so this cation will rearrange
|
3.3 Distinguishing SN1 and SN2 Reactions |

� NOTE: the factors above favor the reactions by making them go faster, e.g. SN2 is FASTER at a primary carbon, SN1 is faster at a tertiary carbon, SN1 is faster in polar protic solvents etc.
� However, weak nucleophiles do not favor SN1 because they make Sn1 reactions faster, they don't, but they do make competing SN2 reactions SLOWER
� SN2 reactions are not precluded by polar protic solvents, they are just faster in aprotic solvents
Examples: assign the mechanism of the following reactions to SN1 or SN2
![]()

Example Problems: Give the major organic product of reactions
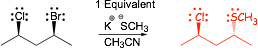
� polar aprotic solvent, strong nucleophile, SN2, Br- better leaving group
� 1 equivalent means exactly the same number of nucleophiles as organic reactants, which in this context means that there is only enough nucleophile to substitute one of the halide leaving groups
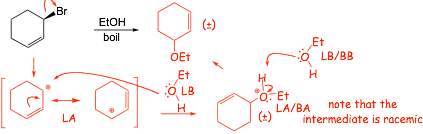
� polar protic solvent and heat, no strong nucleophile and allylic halide, must be SN1. Need to draw the mechanism to be sure of the product!

� polar aprotic solvent, strong nucleophile, SN2, allylic position more reactive
� 1 EQUIVALENT will ONLY REACT at the carbon where SN2 will be fastest
|
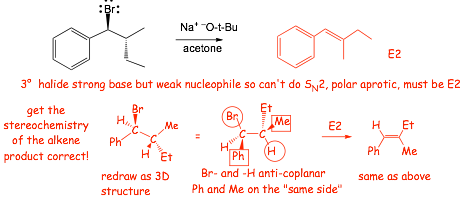
4 E2 Elimination Reaction |
HERE is a reaction that we now know, straightforward SN2 at a primary bromide......

� what about this reaction? SN2 is not possible here (3� bromide), yet there certainly IS a reaction
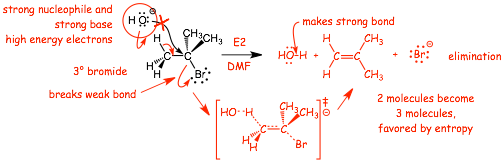
� does E2 instead!!
� breaks weak C�Br bond and strong C�H bond, makes strong O-H bond and C=C pi bond
� not as exothermic as SN2, but the reaction converts 2 molecules into 3, favored by entropy AND "converts" strong -OH base into weak -Br base, this lowers the energy of these electrons, which also helps
� Just like SN2, all bonds made and broken at same time (all four!)
� the �OH acts as a Br�nsted base, a STRONG base is required for E2! What we are doing here is using the chemical potential energy (reactive electrons) in the strong base to "drive" this reaction, to make it "go"
� The reaction is CONCERTED (all FOUR bonds are made and broken at the same time)
4.1 Product Selectivity in E2: Saytzeff Rule (or Zaitsev, etc.) |
� Saytzeff Rule: Most substituted alkene formed, if possible
Example
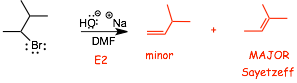
� When more than one alkene isomer can be formed in an E2 elimination, the more substituted, more stable alkene isomer is usually formed (see an exception below), this more substituted alkene is called the Sayetzeff alkene, after the Russian chemist of the same name
� His name was translated differently from the Cyrillic into Roman alphabet in different countries, hence Sayetzeff can be spelled multiple ways, Zaitsev is another common spelling
� consider how these two E2 products are formed
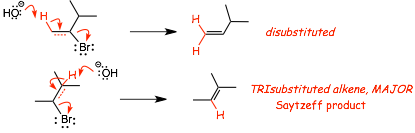
Recall: The most substituted alkene is always the most stable alkene isomer
� the Sayetzeff/Zaitsev alkene is the most substituted of all possible structurally isomeric alkenes that can be formed in an elimination reaction
� E2 eliminations will always form the Sayetzeff/Zaitsev alkene unless there are steric inhibitions, see below
|
4.2 Reactivity Order for E2 |
� decreasing reactivity order: 3� > 2� > 1� halide
Example
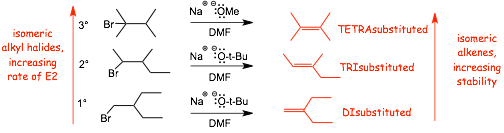
� elimination from a 3� halide tends to give a more substituted alkene product, tends to be faster
� this is one way that we can easily distinguish the possibilities of SN2 versus E2, there is simply no SN2 at a tertiary halide, but E2 eliminations tend to be facile (assuming a strong enough base)
|
4.3 Stereochemistry of E2 Reaction |
� Again, Molecular Orbital theory provide a very informative picture
� We need to make and break ALL FOUR bonds at the same time, the reaction is concerted
� We need to make the new bonds by overlapping the HOMO and the LUMO IN PHASE to make new bonding molecular orbital

� the 2 sigma M.O.s on the central carbons (associated with the breaking C-H and C-Br bonds) become the pi M.O.s (bonding and antibonding)
� the 2 sigma M.O.s therefore must be parallel in order to be able to make the new pi-bond
� the H and leaving group (Br) must be coplanar (periplanar), and preferably "anti"
� the electrons in the breaking C-H sigma bond are used to make the new pi-bond, overlap occurs with the anti-bonding M.O. associated with the breaking C-Br bond best as shown, i.e. analogous to "backside attack" in the SN2 reaction, this is the origin of the requirement for ANTI- in addition to co-planar
� Only One conformation will tend to be reactive in an E2 Reaction
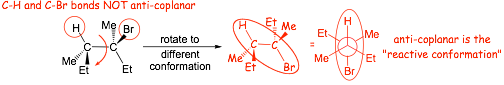
� the reactive conformation must be attained BEFORE reaction can occur
Example: give the major products of the E2 reactions of:
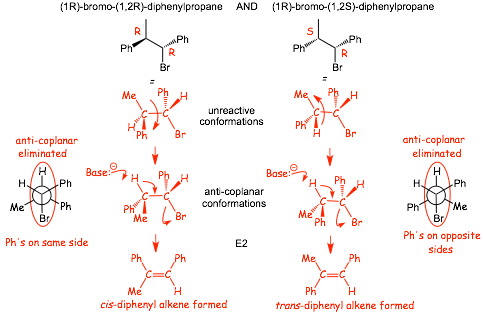
� The reaction is STEREOSPECIFIC, different isomeric halides give different isomeric alkenes because the reaction is CONCERTED
Example: give the major product of the E2 reaction of (1R)-iodo-(2S)-methylcyclohexane
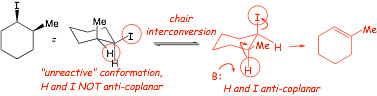
Example: give the major product of the E2 reaction of the following compound
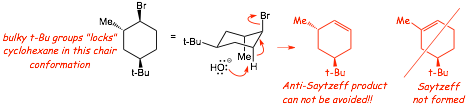
� The cyclohexane is "locked" into the chair that has the LARGE t-butyl substituent in the equatorial position, i.e., the energy difference between the two chairs is so large that the one with the axial t-butyl is present at such low concentration that it can be ignored
� In this conformation, the C-H bond that is anti-coplanar to the C-Br bond MUST give the Anti-Sayetzeff alkene
Example: give the major product of the following reaction......
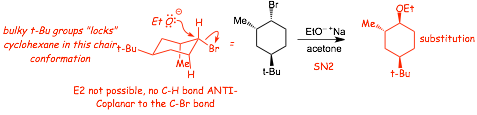
� Again, the Br is "locked" into an equitorial position because of the LARGE t-butyl group
� In this conformation (chair) that are NO anti-coplanar C-H bonds, therefore E2 is NOT POSSIBLE and SN2 is the only reasonable reaction
|
5 E1 Elimination Reaction |
� elimination initiated by 1st-order heterolysis
Example: a 3� halide with a poor nucleophile, poor Br�nsted base and in a polar protic solvent
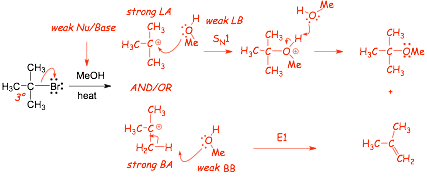
� the intermediate cation is a strong electrophile that can react with the weak nucleophile MeOH via SN1
� the intermediate cation is ALSO a VERY STRONG Br�nsted acid, stronger than hydrochloric acid (pKa < -10)
� The intermediate cation can, therefore, react with the weak Bronsted base MeOH in a Bronsted acid/base reaction to give an alkene ELIMINATION product
� E1 elimination because one molecule is involved in the rate determining step (kinetically first order)
� SN1 and E1 are often competitive, they have the SAME rate determining step, the reactions "partition" at the cation intermediate
� It is difficult to select conditions that favor E1 (high temperature can help due to the temperature dependence of entropy), i.e. not useful "synthesis" reaction - see later
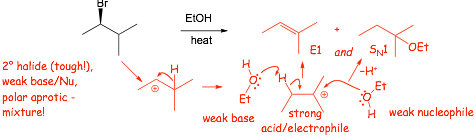
Example: Give the expected ELIMINATION products (ignore substitution) under the following conditions
|
6 Distinguishing E1, E2, SN1 and SN2 Reactions |
� Reality - the mechanisms are often mixed : However, favored conditions are........
SN2 - 1� halide, aprotic solvent, strong nucleophile, weak base
SN1 - 3� halide, protic solvent, weak nucleophile, weak base
E2 - 3� halide, aprotic solvent, weak nucleophile, strong base
E1 - 3� halide, protic solvent..... difficult to favor!
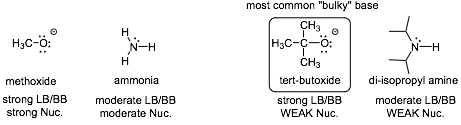
Strong Base but Weak Nucleophile? What is this?

� The tert-butoxide anion makes a strong bond to (for example) a proton, just as strong a bond as the methoxide anion, they are equally strong bases
� However, the t-butoxide anion is not a strong nucleophile, it undergoes SN2 reactions very slowly (remember the definition of nucleophilicity is based on how FAST a reaction is, not how exothermic it is) due to steric interactions associated with the t-butyl group, SN2 reactions are sterically hindered, it is a BULKY BASE
� t-butoxide is a strong bulky base that is a weak nucleophile, this can be used to direct E2 reactions over competitive SN2 reactions, we will see other bulky bases as we work through the course
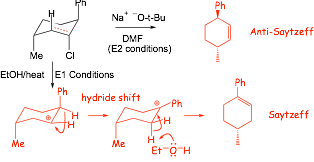
Examples: What was the mechanism that resulted in the PROVIDED organic product? (there may be other reaction products, but the questions ask about the provided ones only)



Example: give the major products of the following reactions and identify the reaction mechanisms

� there is no REQUIREMENT for an SN2 reaction to be in a polar aprotic solvent, they are faster in aprotic solvents but in reality many are actually performed in protic solvents for convenience

� secondary halides don't favor any mechanism in particular and often undergo more than one reaction
� it is usually a good idea to draw out at least a partial mechanism when carbocation intermediates are involved to avoid missing any rearrangements
� SN1 and E1 are often competitive, unless elimination is not possible because there are no adjacent hydrogen atoms
� elimination is not possible in this case
� note the use of the t-butoxide anion BULKY BASE to force E2 elimination'� for E2 eliminations where there is stereochemistry in the reactant, you will usually have to setup the correct conformation for elimination (anti-coplanar) in order to get the correct stereochemistry in the alkene product
|
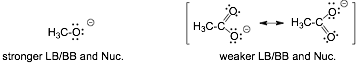
7 Summary of Nucleophiles and Bases |
Most of the time basicity (LB) and nucleophilicity (Nuc) exhibit the same trends that we already understand:
Strong Nucleophiles are also Strong bases because strong Bases make strong bonds
For Example
� Negatively charged species are generally stronger nucleophiles AND bases
![]()
� Basicity and nucleophility decrease with resonance delocalization
� More electronegative elements tend to be weaker nucleophiles AND bases
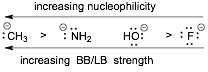
� Basicity AND Nucleophilicity decrease going DOWN the Periodic Table, IN APROTIC SOLVENTS
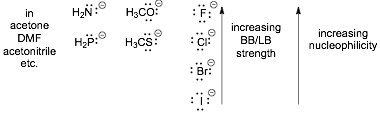
BASICITY and Nucleophilicity are OPPOSITE IN A THREE SITUATIONS
1) Basicity DECREASES but Nucleophilicity INCREASES for ANIONS going down the periodic table IN PROTIC SOLVENTS
� In protic solvents the small anions are highly solvated which decreases their kinetic reactivity and nucleophilicity
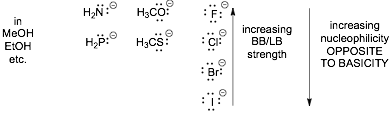
2) Basicity DECREASES but nucleophilicity INCREASES for NEUTRALS going down the periodic table IN ALL SOLVENTS
� Neutrals are not as affected by solvent and the large size and polarizability of the electrons on the larger atom wins out over electronegativity
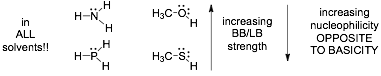
3) Strong bases that are sterically hindered are weak nucleophiles, these are the so-called "bulky" bases
|
8 Elimination Using Bulky (Sterically Hindered) Bases |
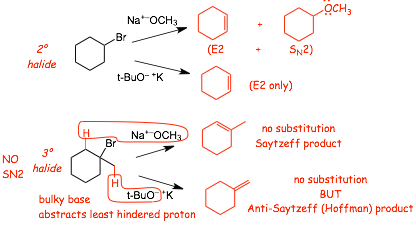
� The products of E2 eliminations can be different for 2 versus 3� halides with or without bulky bases
Examples
Transition states explain product distribution
� the FASTEST reaction occurs, the reaction is KINETICALLY CONTROLLED
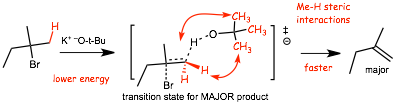
� the transition state for formation of the MAJOR product has CH3-H electron repulsion/steric effects, which costs LESS energy that CH3-CH3 electron repulsions/steric effects below, the transition state is thus lower in energy, the reaction is faster and this reaction forms the MAJOR product
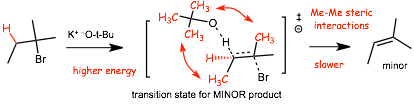
� the transition state for formation of the minor product has CH3-Ch3 electron repulsion/steric effects, which costs MORE energy that CH3-H electron repulsions/steric effects in the reaction that forms the major product, the transition state is thus higher in energy, the reaction is slower and this reaction forms the minor (Hoffman) product
Summary:
� a NON-BULKY with a 3� halide forms the MOST substituted alkene (normal Saytzeff product)
� BUT, a BULKY BASE with a 3� HALIDE forms the LEAST SUBSTITUTED alkene (Hofmann product) for steric reasons
|
9 Water as a Leaving Groups |
� Substitution and elimination reactions are NOT LIMITED to alkyl halides with halide anions as leaving groups
� It is a good idea to look at some other leaving groups now, not to create more stuff to learn, but to facilitate learning of these new reaction types in different contexts
� Good leaving groups are stable as anions
� Leaving groups that are stable NEUTRAL molecules therefore should be expected to be EVEN BETTER!
� A classic example is water, consider the following reaction that converts an alcohol into an alkyl bromide

� This reaction doesn't work! In fact, goes in reverse �OH will substitute for X� (think about a standard SN2 reaction that has -OH as the NUCLEOPHILE and Br- as the Leaving group), this reaction has the OPPOSITE!
��OH is too poor a leaving group, need to make a better leaving group
Consider the following reaction instead
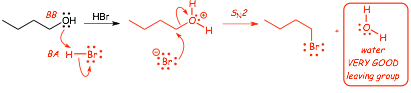
� The first step is a standard Bronsted acid/base reaction, with H-Br as the strong Bronsted acid
� NOW we have a very good potential leaving group, H2O, the next step is standard SN2 and it works well (similar to the tosylate reactions mentioned above) even though the bromide anion is a poor nucleophile
Example Problem:
� Give the mechanism of the following substitution reaction
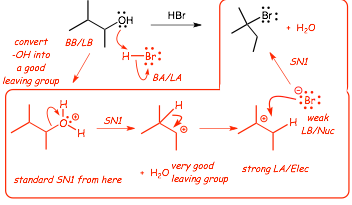
� FIRST, standard Bronsted acid base reaction between the -OH and the strong Bronsted acid H-Br, this converts the -OH from a poor leaving group into a very good leaving group
� After that it is standard SN1 mechanism with an expected rearrangement of the cation intermediate
Elimination of Water
� E1 and E2 eliminations are also observed where water can be a good leaving group
� Again, the reactions start by protonating the -OH of an alcohol to form a good leaving group, and then standard E1 and/or E2 mechanisms after that
Example Problem: Give the product AND Mechanism for the following elimination reaction:
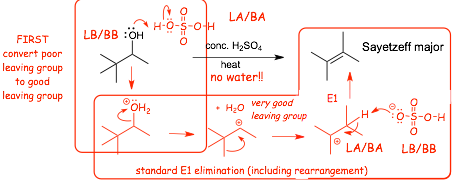
� Sulfuric acid protonates the -OH in a Bronsted acid/base reaction to convert the -OH into a good leaving group
� Water is such a good leaving group that the elimination is almost always E1 with 3� and 2� alcohols
� Water is such a good leaving group that E1 is occurs even at a secondary carbon to make a secondary cation
� carbocation intermediates mean rearrangements! (that hasn't changed, of course)
� the conjugate base anion of the sulfuric acid, the bisulfate anion, is the most likely base to deprotonate the carbocation intermediate, thus regenerating the acid catalyst
� The alkene formed will be the Sayetzeff (Zaitsev), there are no stereochemical constraints in the E1 mechanism and the most stable alkene will form
Why does the alcohol make an alkene + water when previously we learned that water + alkene gives an alcohol?
� THIS is what we learned previously

� The addition reaction "goes" because the weaker pi-bond is converted into a stronger sigma-bond
� The reagents/conditions have a LARGE quantity of water and a SMALL quantity of sulfuric acid
� THIS is what we now learned
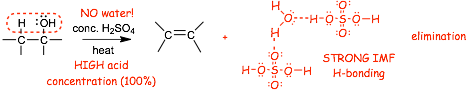
� The reagents have ZERO water and a HIGH concentration of sulfuric acid (opposite of previous reaction)
� The elimination reaction "goes" because the water is highly solvated in the concentrated sulfuric acid
� note a special kind of SOLVENT EFFECT here! In an aqueous medium, acid catalyzes water ADDITION to the alkene to make an alcohol. In conc. sulfuric acid medium, the acid helps to REMOVE water from an alcohol to make an alkene (the sulfuric acid DEHYDRATES the alcohol)
� Alternate reagents and conditions are H2SO4/P2O5, and others�.
Example: Primary (1�) Alcohols: E2 elimination (with rearrangement�)
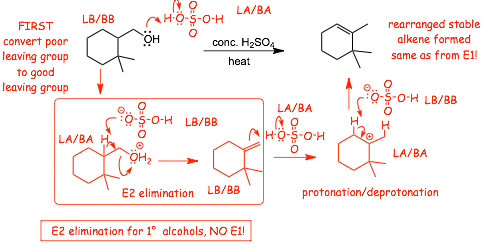
� With a primary alcohol the mechanism must be E2, formation of a primary carbocation CAN'T occur
� BUT, even though the elimination does not involve a rearrangement, the final alkene product is usually the same one that would have been formed via an E1 reaction due to protonation followed by deprotonation (isomerization) of the primary alkene into a final more stable product
Look AGAIN at the second part of the mechanism, the rearrangement
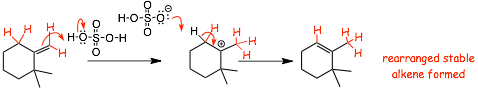
� This effectively converts a less stable less substituted alkene into a more stable more substituted alkene, this is why this ISOMERIZATION reaction "goes"
� To solve the mechanism problem, ADD SOME hydrogen atoms back to the line-angle structure, the H atoms tell you exactly where you need to protonate and deprotonate
� in the presence of acid, PROTONATION will occur first, followed by deprotonation
� a less substituted/less stable alkene is converted into a more substituted/more stable alkene
� this is a REARRANGEMENT, the acid is only the catalyst (no atoms are overall added or subtracted)
� In a strong acid, especially with heat, protonation and deprotonation can OFTEN occur, and if this can result in formation of a more stable alkene, then the more stable alkene will form, and you should always include this step when doing acid catalyzed dehydrations of alcohols
The final product is the SAME MOST SUBSTITUTED ALKENE, whether the mechanism is E1 followed by cation rearrangement (2� and 3� alcohols) or E2 followed by protonation/deprotonation (1� alcohols)
|
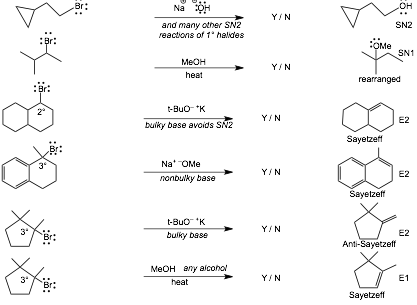
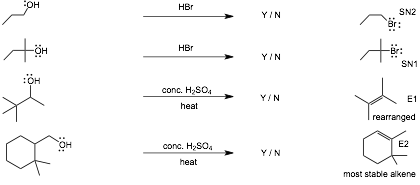
10 Reaction Summary |
Do NOT start studying by trying to memorize these reactions!
Work as many problems as you can with this list of reactions in front of you, if necessary, so that you can get through as many problems as you can without getting stuck on the reagents/conditions. AFTER you have worked all of the problems, just before an exam, then do the following:
� Cover the entire page of reagents/conditions with a long vertical strip of paper, see if you can write down the reagents/conditions for each reaction, check to see which you get correct, if COMPLETELY correct, circle Y, if incorrect or even slightly incorrect, circle N. In this way you keep track of what you know and what you don't know.
� Keep coming back to this list and so the same thing only for those reactions you circled N, until all are circled Y.
� Knowing the reagents/conditions on this page is INSUFFICIENT to do well on an exam since you will ALSO need to recognize how to use and solve reaction problems in different contexts, this page ONLY helps you to learn the reagents/conditions that you have not YET learned by working problems.
ALSO, SN2 reactions in particular can occur in MANY DIFFERENT CONTEXTS, knowing the reactions summarized here is INSUFFICIENT for you to solve substitution and elimination reaction problems